Table Of Content
A similar hit rate was found for the ROCK1 kinase screening in the same study, with one hit in the low nanomolar range26. V-SYNTHES is being applied to other therapeutically relevant targets with well-defined pocket structures. Chemical spaces of gigascale and terrascale, provided that they maintain high drug likeness and diversity, are expected to harbour millions of potential hits and thousands of potential lead series for any target.
3. Artificial Intelligence and Drug Discovery
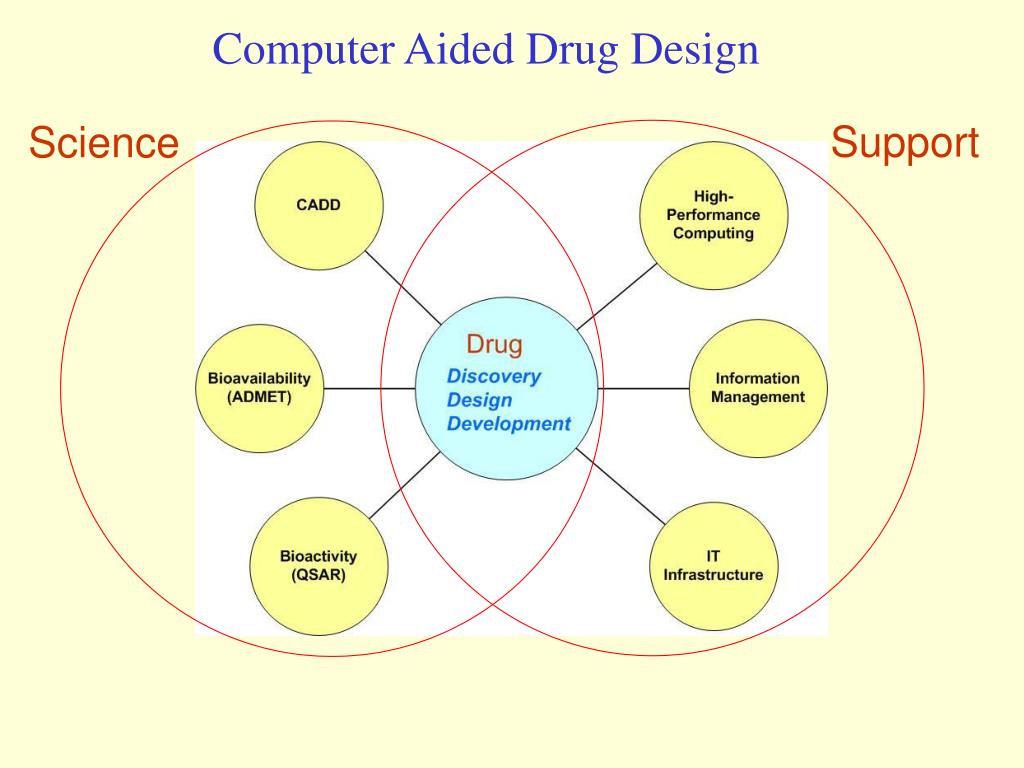
This chapter aims to illustrate some crucial CADD techniques also commonly referred to as in silico methods in a diverse arena of drug discovery and focusing some of the modern advancements. Computer-aided drug discovery has been around for decades, although the past few years have seen a tectonic shift towards embracing computational technologies in both academia and pharma. This shift is largely defined by the flood of data on ligand properties and binding to therapeutic targets and their 3D structures, abundant computing capacities and the advent of on-demand virtual libraries of drug-like small molecules in their billions. Taking full advantage of these resources requires fast computational methods for effective ligand screening.
Meet the UC Campus Leads
Such machines are already working, although scaling-up production of thousands of specialized building blocks remains the bottleneck. The advent of DL takes data-driven models to the next level, allowing analysis of much larger and diverse datasets while deriving more complicated non-linear relationships, with vast literature describing specific DL methodologies and applications to drug discovery27,70. By its ‘learning from examples’ nature, AI requires comprehensive ligand datasets for training the predictive models.
2 Site Identification by Ligand Competitive Saturation (SILCS)
Today, a number of computational approaches are being used to identify potential lead molecules from huge compound libraries. Advancements in science and technology led to the discovery of macromolecules that played a direct or indirect role in a specific disease. In turn, these discoveries guided chemists to design and synthesize novel bioactive compounds increasingly more active than their predecessors. However, getting a druggable compound to market consists of several steps, many challenges and a tremendous amount of work, time and budget.
Data-driven approaches have a long history in drug discovery, in which ML algorithms such as support vector machine, random forest and neural networks have been used extensively to predict ligand properties and on-targets activities, albeit with mixed results. ML is also implemented in many quantitative SAR (QSAR) algorithms75, in which the training set and the resulting models are focused on a given target and a chemical scaffold, helping to guide lead affinity and potency optimization. Methods based on extensive ligand–target binding datasets, chemical similarity clustering and network-based approaches have also been suggested for drug repurposing76,77. This is an important factor in assuring the patentability of the chemical matter for hit compounds and the lead series arising from gigascale screening. Moreover, thousands of easily synthesizable analogues assure extensive SAR-by-catalogue for the best hits, which, for example, enabled approximately 100-fold potency and selectivity improvement for the CB2 V-SYNTHES hits26.
Drug Discovery Market Size to Reach USD 181.4 Billion by 2032 with a CAGR of 8.5% According to Acumen Research ... - GlobeNewswire
Drug Discovery Market Size to Reach USD 181.4 Billion by 2032 with a CAGR of 8.5% According to Acumen Research ....
Posted: Mon, 04 Sep 2023 07:00:00 GMT [source]
Genetics of human brain development
AI methods have been developed to deal with this big data of high volume and multidimensional nature to efficiently predict drug efficacy and side effects in animals or humans. The most promising approach in the present big data world is deep learning which was first used in the drug discovery process in 2012 QSAR machine learning challenge backed by Merck [110]. The results showed that deep learning models were true which can accurately predict the ADMET properties compared to traditional machine learning methods. Although, AI is an impressing method in identification of preclinical candidates in more cost and time-efficient manner, and the accurate prediction of binding affinity between a drug molecule and a receptor using AI remains challenging for quite a several reasons. Firstly, AI is a data mining method whose performance heavily relies on the amount and quality of the available data [4, 111]. Variability in the source of data especially those derived from different biological assays and lack of high-quality data from public databases presents difficulty in efficient AI learning [112, 113].
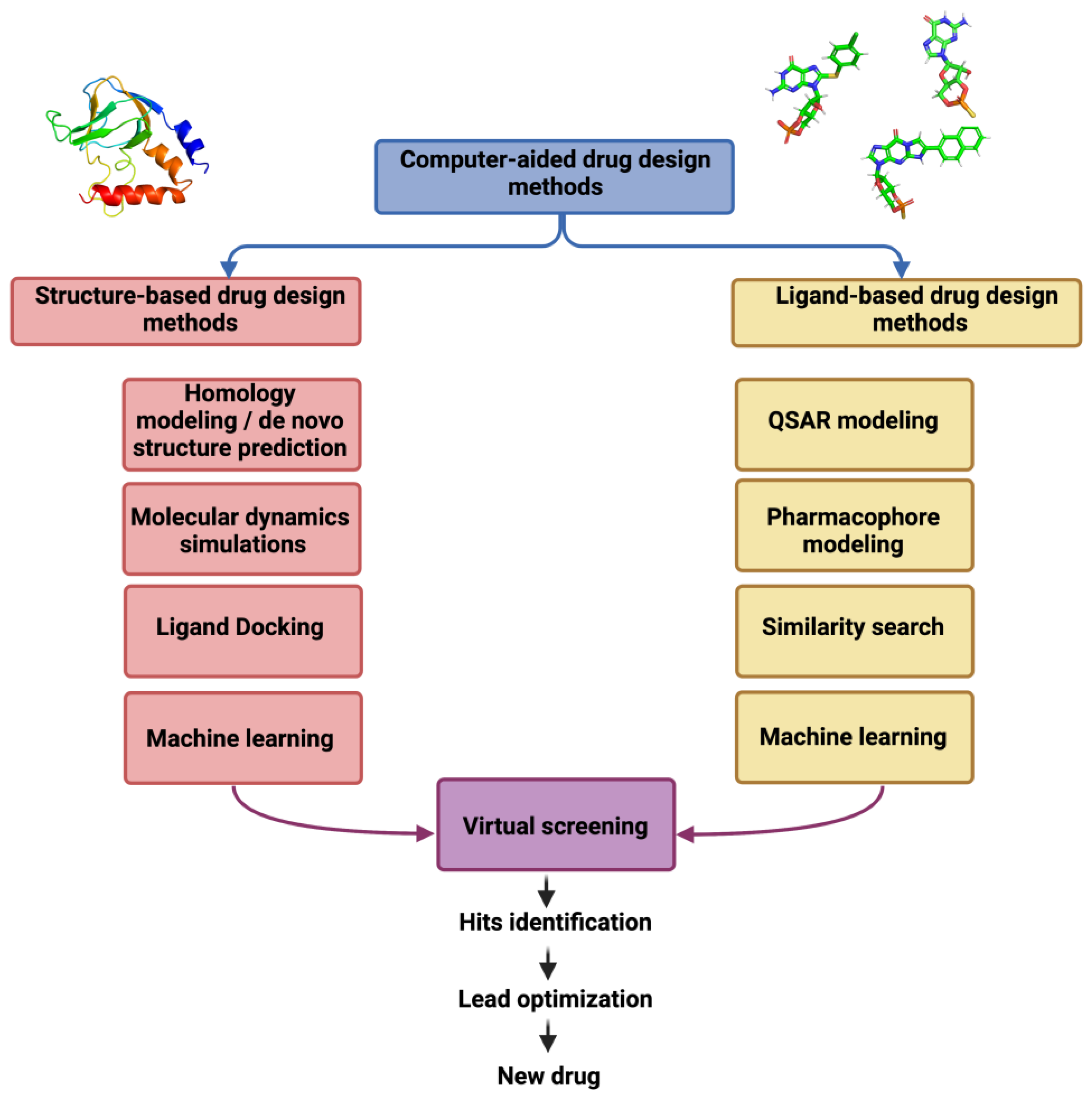
Ligand-based drug design is another widely used approach used in computer-aided drug design and is employed when the three-dimensional structure of the target receptor is not available. The information derived from a set of active compounds against a specific target receptor can be used in the identification of physicochemical and structural properties responsible for the given biological activity which is based on the fact that structural similarities correspond to similar biological functions [77]. Some of the common techniques used in the ligand-based virtual screening approach include pharmacophore modeling, quantitative structure-activity relationships (QSARs), and artificial intelligence (AI).
My comments on ICMJE Data-sharing proposal
Knowledge of the relationship of these properties to activity (i.e. SAR) can be used by the medicinal chemist to qualitatively design new, synthetically-accessible compounds that can be quantitatively evaluated. When developing SAR using pharmacophore descriptors, the appropriate conformations of the compounds that are responsible for the biological activity must be used. Here we illustrate the development of SAR using our in-house developed conformationally sampled pharmacophore (CSP) protocol (94, 95). Docking is a useful CADD tool to predict binding orientation of a ligand molecule within target binding site as well as to evaluate its binding strength (70). Traditional docking methods only consider rigid or limited protein flexibility and ignore or treat the contribution of desolvation to binding in an empirical way.
All the pharmacophoric features which are not consistently detected in active molecules are either made optional or removed from the final model [7]. The pharmacophore model generated should have optimum sensitivity and specificity to minimize the chances of false negative and false positive results and must be validated using an independent external test set [81]. If the information about the 3D structure of a receptor and a set of known active compounds are lacking, then a sequence-derived 3D pharmacophore model is quite useful. For example, Pharma3D utilizes knowledge of the 3D crystal structures and homology models to derive the common sequence motif important for receptor-ligand biomolecular interactions in protein families [81, 82]. During the recent era of drug design, many therapeutically potent and well accepted drug targets with unknown active site geometries have been identified.
In many cases, a combination of several techniques has to be used and methods are frequently adjusted for different drug design projects [8–10]. In many cases, a combination of several techniques has to be used and methods are frequently adjusted for different drug design projects [8,9,10]. The term drug design describes the search of novel compounds with biological activity, on a systematic basis. In its most common form, it involves modification of a known active scaffold or linking known active scaffolds, although de novo drug design (i.e., from scratch) is also possible.
A binding-site identification method under the SILCS framework, named SILCS-Hotspots, was developed recently by our laboratory (115). SILCS-Hotspots is designed to identify fragment binding hotspots that are spatially distributed across the global protein structure including both surface and interior binding sites. The general protocol using SILCS-Hotspots to identify putative binding sites on a protein is described as the following. With the fast development of more powerful computing hardware, expensive algorithms such as free energy perturbation methods (23), which can only be used to finely tune the drug candidates at the lead optimization stage, become much more affordable and have been routinely used in a range of applications (24–26).

Therefore, the good performances of DL models based on PDBbind rely on memorizing similar ligands and receptors, rather than on capturing general information about their binding. One possible explanation for this phenomenon is that the PDBbind database does not have an adequate presentation of ‘negative space’, that is, ligands with suboptimal interaction patterns to enforce the training. The quality of QSAR models, however, differs for different target classes depending on data availability, with the most advances achieved for the kinase superfamily and aminergic GPCRs. An unbiased benchmark of the best ML QSAR models was given by a recent IDG-DREAM Drug-Kinase Binding Prediction Challenge with the participation of more than 200 experts78. The top predictive models in this blind assessment included kernel learning, gradient boosting and DL-based algorithms. The top-performing model (from team Q.E.D) used a kernel regression, protein sequence similarity and affinity values of more than 60,000 compound–kinase pairs between 13,608 compounds and 527 kinases from ChEMBL79 and Drug Target Commons80 databases as the training data.
The main protease is a cysteine protease with a catalytic dyad (cysteine and histidine) in its active pocket [116]. The action of the catalytic activity of Mpro on polyproteins results in the release of the vital proteins required for viral replication by cleaving at least 11 sites around the C-terminal and the central regions of the viral polyproteins with sequence consensus X-(L/F/M)-Q↓(G/A/S)-X [117, 118]. Papain-like protease (PLpro) is the second SARS-CoV-2 proteases potentially targetable with small molecules which cleave three sites, with recognition sequence consensus “LXGG↓XX” [118]. It is an attractive drug target because of its essential role in not only the cleavage and maturation of viral polyproteins and assembly of the replicase-transcriptase complex but also disruption of host immune responses [119]. RNA-dependent RNA polymerase (RdRp) is the cleavage product of the polyproteins 1a and 1ab from ORF1a and ORF1ab and is involved in the replication and transcription of the SARS-CoV-2 genome [120].








No comments:
Post a Comment